
The Food and Drug Administration is unable to review all the reports of problems and injuries tied to medical devices, one of several shortcomings that “raise serious concerns” about the agency’s ability to ensure safety, the Government Accountability Office told a congressional panel today.
The GAO testimony arrives a week after a joint investigation by ProPublica and the Chicago Tribune also found troubles with the FDA’s monitoring of “adverse event” reports in the case of a North Carolina plant that made pre-filled medical syringes.
In late October of 2007, the company shipped tens of thousands of syringes in a lot that was later found to have bacterial contamination. Authorities linked the syringes to 162 injuries and four deaths.
Over several years, reports came in to the FDA’s device division about syringe problems, including complaints of red, white and brown particles in fluid that was supposed to be sterile. The reports came under two different headings: the North Carolina manufacturer’s name, AM2PAT, and the product’s brand name, Sierra Pre-Filled.
Complaints also went to several of the company’s distributors and to the Drug Quality Reporting System. The company also was required to send the FDA reports it deemed valid, although most of those it received were dismissed, according to FDA inspection records.
FDA spokeswoman Siobhan DeLancey told ProPublica earlier that the AM2PAT reports were turning up in several places and that the agency is working on two new data systems that will centralize reporting. At least one of the data systems may be years from completion, a recent GAO report said.
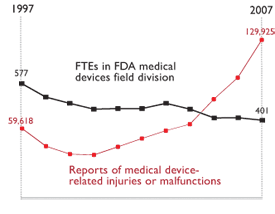 Today’s testimony by Marcia Crosse, the GAO health director, noted that the FDA’s inability to examine all the device-complaint reports makes it tough for regulators to identify and weigh risks. The overall number of adverse-event reports rose nearly fivefold, to 320,000, between 2000 and 2006.
Today’s testimony by Marcia Crosse, the GAO health director, noted that the FDA’s inability to examine all the device-complaint reports makes it tough for regulators to identify and weigh risks. The overall number of adverse-event reports rose nearly fivefold, to 320,000, between 2000 and 2006.
In the case of AM2PAT, FDA officials were aware that a distributor had recalled 1.3 million of the company’s syringes two months before the tainted batch went out the door.
Although the agency’s operations manual says it “should” perform a full inspection to root out problems in such recalls, no such inspection was initiated before the contaminated syringes shipped.
Crosse’s testimony said that GAO reports in 2006 and 2008 found that the FDA couldn’t keep up with medical device complaints. “In 2008, FDA officials told us that while they have a number of strategies to prioritize their reviews, they still cannot review all the reports they receive,” Crosse wrote.
Under the FDA’s system, manufacturers are required to report device-related deaths, serious injuries and certain malfunctions to the agency. User facilities, such as hospitals, must report device-related deaths to the FDA and deaths and serious injuries to the manufacturer.
Consumers and health care professionals may voluntarily report. The information goes into FDA databases for review and safety assessments, which can lead to recalls or public advisories.
In an interview with ProPublica, former FDA official Peter Hutt, now a Harvard law professor, said the agency’s automated information systems fall far short of where they should be. Chronic staffing shortages also hamper the agency’s ability to assess complaints, he said.
“I couldn’t remember a crisis on which we had enough information to make an intelligent decision,” Hutt said. “You just have to decide. That’s the nature of the problem the FDA faces, not every day, but every hour of every day.”
Dr. Allen Roses, chief of neurology at Duke University School of Medicine, worked on a report (PDF) that concluded that the FDA’s information systems are “problematic at best — and at worst it is dangerous.”
Roses said local police departments have systems that show when assaults are starting to occur in one area of town. The FDA, he said, has nothing nearly as sophisticated in monitoring drug or device problems. What’s more, he said that the complaints are often unclear or unreliable.
During the hearing, legislators and experts also presented problems – and examples – of breakdowns in the FDA’s device-approval duties. Crosse testified that complicated devices that should undergo the most rigorous pre-market testing are being fast-tracked.
Rep. Michael Burgess, R-Texas, a physician, noted that the FDA during the 1990s was criticized for approving devices too slowly.”We may have the opposite problem now,” Burgess said, “and none of us should forget that speed sometimes kills.”










